Reaction-diffusion introduction
This is the first in a collection of tutorials about NEURON’s reaction-diffusion module. You may also be interested in these topics:
varying initial concentrations and parameters
non-uniform initial conditions
calcium wave
location-dependent parameters
limiting a reaction to part of a region
radial diffusion
thresholding reactions
3D intracellular
extracellular diffusion
See also:
Reaction-Diffusion exercises from the 2018 NEURON course
Overview
Proteins, ions, etc… in a cell perform signalling functions by moving, reacting with other molecules, or both. In some cases, this movement is by active transport processes, which we do not consider here. If all movement is due to diffusion (wherein a molecule moves randomly), then such systems are known as reaction-diffusion systems.
These problems are characterized by the answers to three questions: (1) Where do the dynamics occur, (2) Who are the actors, and (3) How do they interact?
Math
Reaction-diffusion equations are equations or systems of equations of the form [\frac{\partial u}{\partial t} = \nabla `:nbsphinx-math:cdot (d :nbsphinx-math:nabla u) + f(u, t)] where (u) is the concentration of some state variable. If the diffusion coefficient (D) is constant, (:nbsphinx-math:nabla :nbsphinx-math:cdot (d :nbsphinx-math:nabla u) = d :nbsphinx-math:nabla`^2 u) where (\nabla`^2) is the Laplacian operator. In one-dimensional Cartesian space, (:nbsphinx-math:nabla`^2 u = u_{xx}), while in three-dimensional Cartesian space (:nbsphinx-math:`nabla`^2 u = u_{xx} + u_{yy} + u_{zz}). This form follows from Fick’s law of diffusion and the Divergence theorem and assumes that the diffusion constant (d) is uniform throughout the spatial domain.
## Specification
To describe a reaction-diffusion problem in NEURON, begin by loading the rxd library:
from neuron import rxd
Then answer the three questions: where, who, and how.
Where
We begin by identifying the domain; i.e. where do the dynamics occur? For many electrophysiology simulations, the only relevant domains are the plasma membrane or the volumes immediately adjacent to it on either side, since these are the regions responsible for generating the action potential. Cell biology models, by contrast, have dynamics spanning a more varied set of locations. In addition to the previous three regions, the endoplasmic reticulum (ER), mitochondria, and nuclear envelop often play key roles.
The rxd.Region class is used to describe the domain:
r = rxd.Region(sections, nrn_region=None, geometry=None)
In its simplest usage, rxd.Region simply takes a Python iterable (e.g. a list or a h.SectionList) of h.Section objects. In this case, the domain is the interior of the sections, but the concentrations for any species created on such a domain will only be available through the rxd.Species object and not through HOC or NMODL.
Example: Region on all sections:
r = rxd.Region(h.allsec())
Example: Region on just a few sections:
r = rxd.Region([soma, apical1, apical2])
If the region you are describing coincides with the domain on the immediate interior of the membrane, set nrn_region=’i’, e.g.
r = rxd.Region(h.allsec(), nrn_region='i')
Concentration in these regions increases when a molecule of the species of interest crosses from outside the membrane to the inside via an NMODL or kschan mechanism. If a species on such a region is named ca, then its concentrations can be read and set via the NEURON and NMODL range variable cai.
Also: for the region just outside the membrane (the Frankenhaeuser-Hodgkin space) use nrn_region=’o’. For full 3D extracellular diffusion, define the region with rxd.Extracellular; for more on extracellular diffusion in NEURON, see Newton et al., 2018 or the extracellular diffusion tutorial.
Many alternative geometries are available, including: rxd.membrane, rxd.inside (this is the default), rxd.Shell (used in the radial diffusion example; coming soon), rxd.FractionalVolume (used in the calcium wave example), rxd.FixedCrossSection, and rxd.FixedPerimeter.
Who
Who are the actors? Often they are chemical species (proteins, ions), sometimes they are State variables (such as a gating variable), and other times parameters that vary throughout the domain are key actors. All three of these can be described using the rxd.Species class:
s = rxd.Species(regions=None, d=0, name=None, charge=0, initial=None, atolscale=1)
although we also provide rxd.State and rxd.Parameter for the second and third case, respectively. For now these are exact synonyms of rxd.Species that exist for promoting the clarity of the model code, but this will likely change in the future (so that States can only be changed over time via rxd.Rate objects and that rxd.Parameter objects will no longer occupy space in the integration matrix.)
Note: charge must match the charges specified in NMODL files for the same ion, if any.
The regions parameter is mandatory and is either a single rxd.Region or an iterable of them, and specifies what region(s) contain the species.
Set d= to the diffusion coefficient for your species, if any.
Specify an option for the name= keyword argument to allow these state variables to map to the NEURON/NMODL range variables if the region’s nrn_region is ‘i’ or ‘o’.
Specify initial conditions via the initial= keyword argument. Set that to either a constant or a function that takes a node (defined below, and see also this example).
Example: to have concentrations set to 47 at h.finitialize(init_v):
s = rxd.Species(region, initial=47)
Note: For consistency with the rest of NEURON, the units of concentration are assumed to be in mM. Many cell biology models involve concentrations on the order of μM; calcium is often even smaller. Besides the need to be consistent about units, this has implications for variable step integration which by default has an absolute tolerance of (10^{-3}) or 1 μM. To address this, use atolscale to indicate that the tolerance should be scaled for the corresponding variable; e.g.
ca = rxd.Species(cyt, name='ca', charge=2, atolscale=1e-4)
Note: initial is also a property of the Species/Parameter/State and may be changed at any time, as in:
ca.initial = 1.2345e-3
Warning: Prior to NEURON 7.7, there was a bug in initial support: if initial=None and name=None, then concentration will not be changed at a subsequent h.finitialize(). The intended behavior is that this would reset the concentration to 0.
How
How do they interact? Species interact via one or more chemical reactions. The primary class used to specify reactions is rxd.Reaction:
r = rxd.Reaction(lhs, rhs, rate_f, rate_b=None, regions=None, custom_dynamics=False)
Here lhs and rhs describe the reaction scheme and are expressed using arithmetic sums of integer multiples of a Species. For example, an irreversible reaction to form calcium chloride might be written:
cacl2_reaction = rxd.Reaction(ca + 2 * cl, cacl2, kf)
where ca and cl are rxd.Species instances and kf is the reaction rate. Since custom_dynamics was not specified this is a mass-action reaction and given the reactants kf has units of 1 / (ms μM2). This corresponds to the system of differential equations:
While we can sometimes ignore the reverse reactions due to them having a high energy barrier, the laws of physics imply that all reactions are in fact reversible. A more correct specification of a mass action reaction for calcium chloride would thus include a backward reaction rate kb here in units of 1/ms:
cacl2_reaction = rxd.Reaction(ca + 2 * cl, cacl2, kf, kb)
While mass-action works well for elementary reactions, this is often impractical for modeling intracellular dynamics where there are potentially many elementary reactions driving the observable reaction. In this case, we often turn to phenomenological models, such as Michelis-Menten kinetics or the Hill equation. To indicate these in NEURON, set custom_dynamics=True and specify the forward and backward rates as the corresponding formula, e.g.
enzymatic_reaction = rxd.Reaction(substrate, product, vmax * substrate / (km + substrate),
custom_dynamics=True)
Note that using Michaelis-Menten kinetics for enzymatic reactions is only appropriate under certain conditions, such as that the concentration of enzyme is low relative to the concentration of the substrate.
Note: rxd.Reaction describes a single molecular reaction. That is, if the left hand side involves \(aA + bB + cC\) then \(a\), \(b\), and \(c\) are the stoichiometry coefficients. In particular, this means that a reaction of 2 * ca + 4 * cl goes to 2 * cacl2 is not equivalent to a reaction where ca + 2 * cl goes to cacl2, as the first requires four chloride and two calcium molecules to come together simulatenously before any reaction occurs, which would cause the \([\mathrm{cl}]\) to be raised to the fourth power instead of the second power. For more background, see the Wikipedia article on the rate equation.
Reactions conserve mass. This property is especially important for stochastic simulations. Often, however, one might wish to model a source or a sink (e.g. protein degradation), or describe the dynamics of a state variable that is not a concentration. In this case, use the non-conservative rxd.Rate.
r = rxd.Rate(species, rate, regions=None, membrane_flux=False)
For example, if IP3 is degraded at a rate proportional to its concentration, then
ip3_degradation = rxd.Rate(ip3, -k * ip3)
Then
The effects of multiple rxd.Rate, rxd.Reaction, etc add together.
As with rxd.Reaction, the regions parameter can be used to restrict the action of an rxd.Rate to specific regions.
The previous methods assume that all the reactants exist in the same region. (If they are not all present in a given location, then the reaction does not occur there.) Pumps and channels can allow material to move between compartments. These are implemented in NEURON using an rxd.MultiCompartmentReaction and using square brackets to specify regions. The rate is proportional to the area of the membrane between the regions, which must be specified. e.g. if ca is a species present on the rxd.Region objects er and cyt then a leak compartment across the ER membrane might be implemented as:
leak = rxd.MultiCompartmentReaction(ca[er], ca[cyt], gleak, gleak, membrane=cyt_er_membrane)
This is explored further in the calcium wave example.
Reading the data
NodeList
For a given species s, s.nodes is an rxd.NodeList of all of its nodes. NodeList objects are derived from the Python list, so they offer all the same methods, e.g. one can access the first node
node = s.nodes[0]
or get the total number of nodes
num_nodes = len(s.nodes)
exactly as if s.nodes was a list, but one can also read and set concentrations (concentration) and diffusion constants (diff) for all nodes in the NodeList in a vectorized way, as well as read the volumes (volume), surface areas (surface_area), regions (region), species (species), and normalized positions (x). For example, to get lists of positions (x) and concentrations (y) suitable for plotting from the NodeList nl, use
x = nl.x
y = nl.concentration
Note that normalized positions always lie between 0 and 1, and so if there are multiple sections a more sophisticated approach is necessary.
When assigning concentration or the diffusion constant, one can either set them equal to an iterable with the same length as the NodeList or to a scalar as in
nl.concentration = 47
Calling a NodeList returns a sub-NodeList of nodes satisfying a given restriction. For example,
nl(soma)
returns a NodeList of all Nodes lying in the h.Section referred to by soma. As this is itself a NodeList, we can get a list of the nodes from nl in the soma section belonging to the rxd.Region referred to by er:
nl(soma)(er)
Valid restrictions for now are section objects, region objects, and normalized position \(0 \le x \le 1\). Restrictions are implemented via the satisfies method of individual nodes. If you would like to extend the set of restrictions, send us a pull request on the NEURON source at GitHub.
Node
Sometimes though, it is important to work with an individual Node, such as for communication with nmodl or for plotting. NEURON’s Graph.addvar() method, for example, needs a reference to the memory location containing the concentration, which is available via the _ref_concentration property:
g.addvar('calcium', node._ref_concentration)
If a node is mapped to a NEURON range variable (that is, if the species name is specified and the Region’s nrn_region is ‘i’ or ‘o’), then there are three equivalent ways to read the concentration in 1d simulations. Here we assume the species is named ca and the nrn_region=’i’ and that the node is at 0.5 in the h.Section soma:
soma(0.5).cai
node.concentration
node._ref_concentration[0]
The latter two can also be used to set concentrations, but if you use this with the variable step solver, you must reinitialize via: h.CVode().re_init()
In 1d, all three will always report the same value. In 3d, however, the values in the NEURON range variables are averaged values, and changing those will not change the 3d values.
All the properties of NodeList are also available in individual Nodes, with the difference being that all values are scalars and not vectors.
Examples
Simple Reaction with Abrupt Change in Reaction Rate
[1]:
from neuron import h, rxd
dend = h.Section(name="dend")
cyt = rxd.Region(h.allsec(), nrn_region="i")
cl = rxd.Species(cyt, initial=1, name="cl", charge=-1)
ca = rxd.Species(cyt, initial=1, name="ca", charge=2)
cacl2 = rxd.Species(cyt, initial=0, name="cacl2")
reaction = rxd.Reaction(2 * cl + ca, cacl2, 1)
h.finitialize(-65)
heading = "{t:>10s} {cl:>10s} {ca:>10s} {cacl2:>10s}"
data = "{t:10g} {cl:10g} {ca:10g} {cacl2:10g}"
def advance_a_bit():
for i in range(5):
h.fadvance()
print(
data.format(
t=h.t,
cl=cl.nodes[0].concentration,
ca=ca.nodes[0].concentration,
cacl2=cacl2.nodes[0].concentration,
)
)
print(heading.format(t="t", cl="cl", ca="ca", cacl2="CaCl2"))
print(heading.format(t="-", cl="--------", cacl2="--------", ca="--------"))
advance_a_bit()
# increase the forward reaction rate
reaction.f_rate *= 5
print()
print("---- cacl2 production rate should speed up below here ----")
print()
advance_a_bit()
t cl ca CaCl2
- -------- -------- --------
0.025 0.955556 0.977778 0.0222222
0.05 0.915565 0.957783 0.0422175
0.075 0.879356 0.939678 0.0603222
0.1 0.846386 0.923193 0.0768069
0.125 0.816217 0.908108 0.0918917
---- cacl2 production rate should speed up below here ----
0.15 0.691965 0.845982 0.154018
0.175 0.602562 0.801281 0.198719
0.2 0.535061 0.76753 0.23247
0.225 0.482188 0.741094 0.258906
0.25 0.43957 0.719785 0.280215
Scalar Bistable Wave
Propagating waves are a form of regenerative signaling that allows messages to travel faster than is possible by diffusion alone; examples in neurons include action potential propagation and calcium waves.
We consider the scalar bistable equation
so named because it requires only a single state variable \(u\) and has two stable solutions \(u=0\) and \(u=1\). This formula reproduces the phenomena of a propagating wave front but it is not based on any specific biological model. Its main advantage is that the exact analytic solution is known on the real line.
[2]:
%matplotlib inline
from neuron import h, rxd
import numpy
from matplotlib import pyplot
# needed for standard run system
h.load_file("stdrun.hoc")
dend = h.Section(name="dend")
dend.nseg = 101
# initial conditions
def my_initial(node):
return 1 if node.x < 0.2 else 0
# WHERE the dynamics will take place
where = rxd.Region([dend])
# WHO the actors are
u = rxd.Species(where, d=1, initial=my_initial)
# HOW they act
bistable_reaction = rxd.Rate(u, -u * (1 - u) * (0.3 - u))
# initial conditions
h.finitialize(-65)
def plot_it(color="k"):
y = u.nodes.concentration
x = u.nodes.x
# convert x from normalized position to microns
x = dend.L * numpy.array(x)
pyplot.plot(x, y, color)
plot_it("r")
for i in range(1, 5):
h.continuerun(i * 25)
plot_it()
pyplot.xlabel("t (ms)")
pyplot.ylabel("[u] (mM)")
pyplot.show()
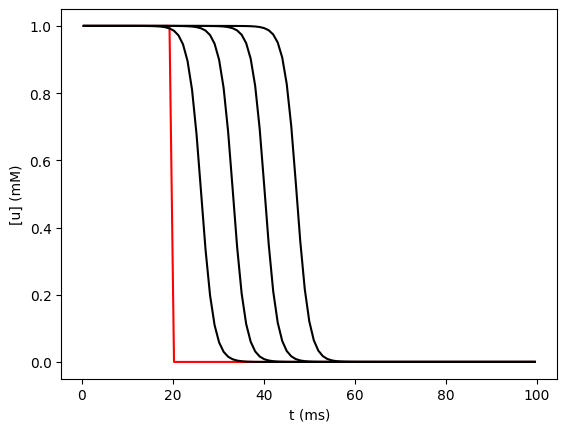
We could, of course, have plotted with any of a number of other graphics libraries, including NEURON’s built in Graph, Bokeh, plotnine, etc.
Besides the graph, this illustrates two other features not shown in the previous example: (1) we explicitly specify the list of h.Section objects for the rxd.Region and (2) we assign non-uniform initial conditions by passing a function that takes a node and returns a value to the initial keyword argument for the rxd.Species.